Zbirke Državnega zbora RS - sprejeti zakoni |
 EVIDENČNI PODATKI
EVIDENČNI PODATKI
Z A K O N
O RATIFIKACIJI EVROPSKEGA SPORAZUMA O IZMENJAVI ZDRAVILNIH UČINKOVIN ČLOVEŠKEGA IZVORA, PROTOKOLA K EVROPSKEMU SPORAZUMU O IZMENJAVI ZDRAVILNIH UČINKOVIN ČLOVEŠKEGA IZVORA TER DODATNEGA PROTOKOLA K EVROPSKEMU SPORAZUMU O IZMENJAVI ZDRAVILNIH UČINKOVIN ČLOVEŠKEGA IZVORA (MESZUC)
1. člen
Ratificirajo se Evropski sporazum o izmenjavi zdravilnih učinkovin človeškega izvora, sklenjen v Parizu 15. decembra 1958, Protokol k Evropskemu sporazumu o izmenjavi zdravilnih učinkovin človeškega izvora, sklenjen v Strasbourgu 19. aprila 1982, ter Dodatni protokol k Evropskemu sporazumu o izmenjavi zdravilnih učinkovin človeškega izvora, sklenjen v Strasbourgu 29. septembra 1982 in dan na voljo za sprejetje 1. januarja 1983.
2. člen
Sporazum, protokol in dodatni protokol se v izvirniku v angleškem jeziku in v prevodu v slovenskem jeziku glasijo:
Evropski sporazum
o izmenjavi
zdravilnih učinkovin
človeškega izvora
Uvod
Vlade podpisnice tega sporazuma, članice Sveta Evrope, so se
glede na to, da zdravilne učinkovine človeškega izvora že po svoji naravi izhajajo iz dejanja človeškega darovalca in torej niso na voljo v neomejenih količinah,
glede na to, da je nadvse zaželeno, da si države članice v duhu evropske solidarnosti med seboj pomagajo pri oskrbi s temi zdravilnimi učinkovinami, če se pokaže potreba po njih,
glede na to, da je taka medsebojna pomoč možna le, če glede lastnosti in uporabe teh zdravilnih
učinkovin veljajo pravila, ki jih skupaj določijo države članice, in če se pri uvozu odobrijo ustrezne olajšave in oprostitve,
sporazumele o naslednjem:
1. člen
V tem sporazumu se izraz »zdravilne učinkovine človeškega izvora« nanaša na človeško kri in njene pripravke.
Določbe tega sporazuma se lahko z izmenjavo pisem med dvema ali več pogodbenicami razširijo na druge zdravilne učinkovine človeškega izvora.
2. člen
Pogodbenice se zavezujejo, da dajo zdravilne učinkovine človeškega izvora na voljo drugim pogodbenicam, ki jih nujno potrebujejo, če imajo dovolj zalog za lastne potrebe, in da zaračunajo le stroške zbiranja, predelave in prevoza teh učinkovin.
3. člen
Zdravilne učinkovine človeškega izvora se dajo drugim pogodbenicam na voljo pod izrecnim pogojem, da z njimi ne ustvarjajo dobička, da jih uporabljajo izključno v medicinske namene in da jih dobavljajo le organom, ki jih določijo njihove vlade.
4. člen
Pogodbenice potrjujejo, da upoštevajo minimalne zahteve glede lastnosti zdravilnih učinkovin in predpise o označevanju, pakiranju in pošiljanju, kot so določeni v protokolu k temu sporazumu.
Izpolnjujejo tudi pravila, ki so jih sprejele glede na mednarodno standardizacijo na tem področju.
Vsem pošiljkam zdravilnih učinkovin človeškega izvora mora biti priložen certifikat, ki potrjuje, da so bile pripravljene v skladu s specifikacijami v protokolu. Ta certifikat je narejen po vzorcu iz Priloge 1 k protokolu.
Protokol in njegove priloge lahko vlade pogodbenic tega sporazuma spremenijo ali dopolnijo.
5. člen
Pogodbenice ukrenejo vse potrebno, da so zdravilne učinkovine človeškega izvora, ki jim jih dajo na voljo druge pogodbenice, oproščene vseh uvoznih dajatev.
Prav tako ukrenejo vse potrebno, da omogočijo hitro dobavo teh učinkovin po najkrajši poti prejemnikom, omenjenim v 3. členu tega sporazuma.
6. člen
Pogodbenice si po generalnem sekretarju Sveta Evrope pošljejo seznam organov, pooblaščenih za izdajo certifikatov, določenih v 4. členu tega sporazuma.
Prav tako pošljejo seznam organov, pooblaščenih za distribucijo uvoženih zdravilnih učinkovin človeškega izvora.
7. člen
Ta sporazum je na voljo za podpis članicam Sveta Evrope, ki lahko postanejo njegove pogodbenice s:
a) podpisom brez pridržka glede ratifikacije ali
b) podpisom s pridržkom glede ratifikacije, ki mu sledi ratifikacija.
Listine o ratifikaciji se deponirajo pri generalnem sekretarju Sveta Evrope.
8. člen
Ta sporazum začne veljati prvi dan v mesecu, ki sledi dnevu, ko tri članice Sveta v skladu s 7. členom tega sporazuma podpišejo sporazum brez pridržka glede ratifikacije ali ga ratificirajo.
Za vsako drugo članico Sveta, ki pozneje podpiše sporazum brez pridržka glede ratifikacije ali ga ratificira, začne sporazum veljati prvi dan v mesecu, ki sledi temu podpisu ali deponiranju listine o ratifikaciji.
9. člen
Odbor ministrov Sveta Evrope lahko povabi katero koli državo nečlanico, da pristopi k temu sporazumu. Pristop začne veljati prvi dan v mesecu po deponiranju listine o pristopu pri generalnem sekretarju Sveta Evrope.
10. člen
Generalni sekretar Sveta Evrope uradno obvesti članice Sveta in države, ki so pristopile k sporazumu, o:
a) datumu začetka veljavnosti tega sporazuma in imenih tistih članic, ki so ga podpisale brez pridržka glede ratifikacije ali so ga ratificirale,
b) deponiranju vsake listine o pristopu v skladu z 9. členom,
c) vsakem uradnem obvestilu, prejetem na podlagi 11. člena, in datumu začetka njegove veljavnosti,
d) vsaki spremembi protokola ali njegovih prilog po četrtem odstavku 4. člena.
11. člen
Ta sporazum velja nedoločen čas.
Vsaka pogodbenica lahko preneha uporabljati sporazum eno leto po uradnem obvestilu generalnemu sekretarju Sveta Evrope.
V potrditev tega so podpisani, ki so jih njihove vlade za to pravilno pooblastile, podpisali ta sporazum.
Sestavljeno v Parizu 15. decembra 1958 v angleškem in francoskem jeziku, pri čemer sta besedili enako verodostojni, v enem izvodu, ki se hrani v arhivu Sveta Evrope. Generalni sekretar pošlje overjene kopije vsem vladam podpisnic in vladam tistih držav, ki pristopijo k temu sporazumu.
PROTOKOL
K EVROPSKEMU SPORAZUMU
O IZMENJAVI ZDRAVILNIH UČINKOVIN ČLOVEŠKEGA IZVORA
___________________
I. DEL
SPLOŠNE DOLOČBE
A. Označevanje
Etiketa, natisnjena v angleščini in francoščini, narejena po ustreznem vzorcu, ki je v prilogah od 2 do 10 tega protokola, je nalepljena na vsak vsebnik ali set za dajanje.
B. Pakiranje in pošiljanje
Polno človeško kri pošiljamo v vsebnikih, v katerih je temperatura ves čas transporta od 4 C do 6 C.
Ta pogoj ni predpisan za pripravke, navedene v protokolu.
C. Pripravki in oprema
Pripravki in oprema, ki so navedeni v drugem delu tega protokola, so sterilni, apirogeni in netoksični.
Priporočamo, da se vsaki pošiljki priložijo set za dajanje kot tudi predpisana topila za dehidrirane pripravke.
D. Netoksičnost plastične opreme za transfuzijo krvi
Oprema je v skladu z določbami, določenimi v prilogi 11 tega protokola.
II. DEL
POSEBNE DOLOČBE
1. Polna človeška kri
Polna človeška kri je kri, ki je mešana z ustreznim antikoagulantom po odvzemu zdravemu krvodajalcu.
Krvi ne odvzamemo krvodajalcu:
a) za katerega vemo, da ima ali je prebolel sifilis ali hepatitis,
b) čigar krvni testi na okužbo s sifilisom niso bili negativni,
c) ki ima, kolikor je mogoče ugotoviti z zdravniškim pregledom in iz podatkov
o boleznih v družini, bolezen, ki je prenosljiva s transfuzijo krvi.
Odvzem opravimo v aseptičnih pogojih s pomočjo zaprtega sistema sterilnih cevk v sterilni vsebnik, v katerega smo pred sterilizacijo dali antikoagulantno raztopino. Uporabljena oprema mora biti apirogena. Po končanem odvzemu vsebnik nemudoma zapremo in ohladimo na 4 C do 6 C; ne smemo ga odpirati do trenutka, ko bo kri uporabljena.
Kri zbiramo v citratni raztopini kisle reakcije, ki vsebuje dekstrozo. Ne dodajamo antiseptičnih ali bakteriostatičnih učinkovin. Volumen antikoagulantne raztopine ne sme presegati 220 ml /l polne človeške krvi in koncentracija hemoglobina ne sme biti manjša od 97 g/l.
Krvna skupina – Krvno skupino po sistemu AB0 je treba določiti s pregledom krvnih celic in seruma, krvno skupino Rh sistema pa s pregledom krvnih celic iz ločenega vzorca krvi krvodajalca. Kadar obstaja neki državni standard ali metoda za določanje krvnih skupin, ki jo priporoča država, uporabimo priporočeno metodo.
Izraz Rh negativen se uporablja le, kadar so posebni testi pokazali odsotnost antigenov C, D, Du in E. Vso drugo kri je treba označiti kot Rh pozitivno.
Kri, izmenjana po tem sporazumu, se sme uporabljati le za prejemnike, ki imajo ustrezno krvno skupino AB0.
Shranjevanje – Do uporabe polno človeško kri hranimo pri temperaturi od 4 C do
6 C v sterilnem vsebniku, ki mora biti zaprt tako, da se prepreči prodiranje mikroorganizmov; v času, ki je potreben za pregled in transport, temperatura sme biti višja do trideset minut, ko je treba kri nemudoma ponovno ohladiti na 4 C do 6 C.
Označevanje – Na etiketi vsebnika so navedeni vsi podatki, prikazani na vzorčni etiketi (Priloga 2). Skupina Rhesus je navedena kot "pozitivna" ali "negativna" ali skrajšano "POZ" ali "NEG".
1 bis Koncentrirani človeški eritrociti
Koncentrirani človeški eritrociti so enota polne človeške krvi, iz katere je odstranjena večina plazme.
Vsebujejo večino eritrocitov enote, iz katere je bil koncentrat pripravljen; prisotne so lahko tudi druge celične sestavine, lahko pa so delno odstranjene.
Tekoča vsebina koncentrata sestoji bodisi iz preostale plazme ali iz ustrezne izotonične ohranitvene raztopine, dodane po odstranitvi plazme. Volumen eritrocitov naj predstavlja od 65 % do 75 % celotnega volumna pripravka, ob višji koncentraciji eritrocitov se na etiketi navede približen odstotek njihovega volumna (hematokrit).
Vse zahtevane postopke priprave izvajamo v aseptičnih pogojih: za prelivanje uporabljamo sterilen, zaprt sistem z uporabo stiskalnikov. Ne smemo dodati nobenih antiseptičnih ali bakteriostatičnih sredstev.
Krvna skupina in shranjevanje – kot za polno človeško kri.
Označevanje – Na etiketi vsebnika so navedeni vsi podatki, prikazani na vzorčni etiketi (Priloga 2 bis). Skupina Rhesus je navedena kot "pozitivna" ali "negativna" ali skrajšano "POZ" ali "NEG". Če je bila dodana ohranitvena raztopina, na etiketi navedemo njen volumen in sestavo.
2. Dehidrirana človeška plazma
Dehidrirano človeško plazmo pripravljamo s sušenjem plazme, ki jo iz polne človeške krvi ločimo s centrifugiranjem ali sedimentacijo.
Med pripravo ne dodajamo nobenih antiseptičnih ali bakteriostatičnih ali drugih učinkovin. Dehidrirano človeško plazmo pridobivamo z liofilizacijo ali s katero koli drugo metodo, s katero se izognemo denaturaciji proteinov. Dehidrirani pripravek je dobro topen v količini vode, ki je enaka volumnu tekočine, iz katere je bila učinkovina pripravljena. Tako pridobljena koncentracija proteinov v raztopini mora biti najmanj 45 g/l; ne sme kazati prisotnosti produktov hemolize. Titer hemaglutininov ni večji od 1 : 32.
Dehidrirana človeška plazma, pripravljena iz ene ali dveh enot
Enote, ki vsebujejo nevarne stopnje izohemolizinov (določenih iz vzorca svežega seruma) ali katerih koli imunohemaglutininov, izločimo. Če plazme ne mešamo in ne zamrznemo v 48 urah po zbiranju krvi, testiramo sterilnost vsake enote z bakteriološko kulturo najmanj 10 ml.
Dehidrirana človeška plazma, pripravljena iz mešanice več kot dveh enot krvi
Mešanice, ki kažejo prisotnost nevarnih stopenj imunohemaglutininov ali izohemolizinov, izločimo. Da se izognemo neugodnim posledicam bakterijskih produktov v plazmi, ne uporabimo nobenih posameznih enot, v katerih dokažemo bakterijsko kontaminacijo; sterilnost vsake mešanice testiramo z bakteriološko kulturo najmanj
10 ml. Da zmanjšamo tveganje prenosa serumskega hepatitisa, plazmo pripravimo iz mešanic, ki naj vsebujejo največ dvanajst enot, ali s katero koli drugo metodo, za katero je dokazano, da na primerljiv način zmanjša tveganje.
Topnost v vodi – Dodamo količino vode, enako volumnu tekočine, iz katere je bil pripravljen vzorec; učinkovina se popolnoma raztopi v 10 minutah pri 15 C do
20 C.
Identifikacija – Raztopimo znano količino pripravka v volumnu vode, ki je enak volumnu tekočine, iz katere je bil pripravljen; raztopino testiramo z naslednjima testoma:
i) precipitacijski test s specifičnimi antiserumi mora pokazati le vsebnost proteinov človeške plazme,
ii) 1 ml dodamo ustrezno količino trombina ali kalcijevega klorida; nastopi koagulacija, ki jo je mogoče pospešiti z inkubacijo pri 37 C.
Izguba mase pri dehidraciji – Dehidrirana človeška plazma ne sme izgubiti več kot 0,5 % svoje mase po dodatnem 24-urnim sušenju ob prisotnosti fosforjevega pentoksida pri tlaku, ki ne presega 0,02 mm živega srebra.
Sterilnost – Po rekonstituciji mora biti končni pripravek sterilen, ko ga preizkusimo z ustrezno bakteriološko metodo.
Shranjevanje – Dehidrirano človeško plazmo moramo hraniti v atmosferi dušika ali v vakuumu v sterilnem vsebniku, ki mora biti zaprt tako, da se prepreči prodiranje mikroorganizmov, in kolikor je mogoče, vlage; zaščiten mora biti pred svetlobo in shranjen pri temperaturi pod 20 C.
Označevanje – Na etiketi vsebnika so navedeni vsi podatki, ki so prikazani na vzorčni etiketi (Priloga 3).
3. Humani albumin in proteinska frakcija človeške plazme
Humani albumin in proteinska frakcija človeške plazme sta pripravka tiste proteinske sestavine, ki tvori približno 60 % celotne proteinske mase v plazmi polne človeške krvi.
Uporabljena metoda priprave je takšna, da dobimo snov, ki ustreza zahtevam, predpisanim v tem protokolu. Ne glede na to, ali je končni pripravek tekoč ali dehidriran, moramo pripravek, potem ko smo mu dodali ustrezno sredstvo ali sredstva za stabilizacijo, 10 ur segrevati v tekočem stanju v končnem vsebniku pri 60 C
0,5 C, da inaktiviramo povzročitelja serumskega hepatitisa. Med pripravo ne dodamo nobene antiseptične ali bakteriostatične učinkovine.
V pripravkih humanega albumina je tega najmanj 95 % mase prisotnih proteinov. V pripravkih proteinske frakcije človeške plazme je albumina najmanj 85 % proteinske mase. V obeh pripravkih je več kot 10 mg imunoglobulina G na gram pripravka.
Kadar končni pripravek liofiliziramo, mora vsebovati najmanj 950 mg proteina na gram pripravka.
Kadar proteinsko frakcijo človeške plazme pripravljamo kot raztopino, je skupna koncentracija proteinov od 45 do 50 g/l.
Kadar humani albumin pripravljamo kot raztopino, je skupna koncentracija proteinov najmanj 45 g/l.
Topnost dehidriranega produkta – Dodamo vodo do oznake; dehidrirani pripravek mora biti popolnoma topen.
Stabilnost – S primerjavo raztopin pred toplotno obdelavo in po njej ne ugotovimo znakov denaturacije proteinov v raztopini, kar lahko ocenimo z meritvami viskoznosti in motnosti, ultracentrifugiranjem in elektroforezo. Po segrevanju pri 57 C in po 6-urnem tresenju v mehaničnem stresalniku pri tej temperaturi je raztopina brez vidnih delcev.
Identifikacija
i) Precipitacijski testi s specifičnimi antiserumi morajo v obeh pripravkih pokazati le vsebnost proteinov človeške plazme.
ii) Pri elektroforezi, ki temelji na gibanju proteinov pod določenimi pogoji, moramo dokazati, da proteinska frakcija z gibljivostjo albuminske sestavine normalne človeške plazme ni manjša od 95 % proteinske mase v pripravkih humanega albumina ali ni manjša od 85 % proteinske mase v pripravkih proteinske frakcije človeške plazme.
Vsebnost natrija in koncentracija natrija – Vsebnost natrija v humanem albuminu, v katerem sol skoraj ni prisotna, ne sme presegati 0,61 mM/g albumina. V drugih pripravkih humanega albumina in proteinske frakcije človeške plazme koncentracija natrija ne sme presegati 0,15 M raztopine ali rekonstituiranega dehidriranega pripravka.
Koncentracija kalija – Koncentracija kalija v proteinski frakciji človeške plazme ne sme presegati 2 mM raztopine ali rekonstituiranega dehidriranega pripravka.
Kislost – pH enega ali drugega pripravka mora biti 6,8 0,2, kadar ga merimo pri temperaturi 15 C do 25 C v raztopini, razredčeni na koncentracijo 10 g proteina na liter s pomočjo raztopine, ki vsebuje 0,15 M natrijevega klorida.
Izguba mase pri dehidraciji – Dehidrirani pripravki ne smejo izgubiti več kot
0,5 % svoje mase po dodatnem 24-urnem sušenju ob prisotnosti fosforjevega pentoksida pri tlaku, ki ne presega 0,02 mm živega srebra.
Sterilnost – Končni pripravek je sterilen, ko ga preizkusimo z ustrezno bakteriološko metodo.
Shranjevanje – Dehidrirani humani albumin moramo hraniti v atmosferi dušika ali v vakuumu v sterilnem vsebniku, ki je zaprt tako, da se prepreči prodiranje mikroorganizmov, in kolikor je mogoče, vlage, zaščiten mora biti pred svetlobo in shranjen pri temperaturi pod 20 C.
Raztopine humanega albumina in proteinske frakcije človeške plazme moramo hraniti v sterilnih vsebnikih, ki morajo biti zaprti tako, da se prepreči prodiranje mikroorganizmov, zaščiteni morajo biti pred svetlobo in shranjeni pri temperaturi od
4 C do 6 C.
Označevanje – Na etiketi vsebnika so navedeni vsi podatki, ki so prikazani na ustrezni vzorčni etiketi (Priloga 4). Datum priprave raztopine je datum toplotne obdelave v končnem vsebniku.
4. Humani imunoglobulin
Humani imunoglobulin je pripravek iz proteinov plazme, pripravljene iz polne človeške krvi, ki vsebuje protitelesa zdravih odraslih ljudi. Pridobivamo ga iz mešanice tekoče človeške plazme najmanj 1000 krvodajalcev.
Uporabljena metoda priprave mora biti taka, da dobimo snov, ki ustreza zahtevam, predpisanim v tem protokolu, in ki preprečuje prenos serumskega hepatitisa s končnim pripravkom. Metoda za pripravo pripravka mora biti tudi taka, da so protitelesa, ki jih vsebuje začetna snov, skoncentrirana v ustrezni količini v končnem pripravku. Vsak postopek za končni pripravek se dokaže kot zadovoljiv tako, da titriramo protitelesa v začetni snovi in v končnem pripravku z najmanj enim virusom in enim bakterijskim toksinom. Izberemo protitelesa, za katera že obstajajo priznane metode titracije.
Med pripravo ne dodamo nobene antiseptične ali bakteriostatične učinkovine; končnemu pripravku lahko dodamo ustrezen konzervans in sredstvo za stabilizacijo, da ohranimo bakterijsko sterilnost in stabilnost končnega pripravka.
Končni pripravek je na voljo kot raztopina, v kateri je koncentracija imunoglobulina od 100 do 170 g/l.
Identifikacija
i) Precipitacijski testi s specifičnimi antiserumi morajo pokazati le vsebnost proteinov
človeške plazme.
ii) Pri elektroforezi, ki temelji na gibanju proteinov pod določenimi pogoji, ima najmanj 90 % mase proteinov gibljivost gama sestavine globulinov normalne človeške plazme.
Stabilnost – Pred 7-dnevnim segrevanjem končne raztopine pri 37 C in po njem ne sme biti znakov precipitacije in motnosti. Priporočljivo je tudi izvesti teste s pomočjo metode ultracentrifugiranja, da določimo, do katere mere pripravek razpade na sestavine nižje molekularne teže. Uporabljeno metodo mora odobriti državni kontrolni organ.
Kislost – pH končne raztopine mora biti 6,8 0,4, kadar ga merimo pri temperaturi 15 C do 25 C, v raztopini, razredčeni na koncentracijo 10 g/l proteinov z raztopino, ki vsebuje 0,15 M natrijevega klorida.
Sterilnost – Končni pripravek je sterilen, ko ga preizkusimo z ustrezno bakteriološko metodo.
Shranjevanje – Raztopino humanega imunoglobulina moramo hraniti v sterilnem vsebniku, ki je zaprt tako, da se prepreči prodiranje mikroorganizmov, zaščiten mora biti pred svetlobo in shranjen pri temperaturi od 4 C do 6 C.
Označevanje – Na etiketi vsebnika so navedeni vsi podatki, ki so prikazani na vzorčni etiketi (Priloga 5). Datum priprave je datum polnjenja končnega vsebnika.
5. Specifični humani imunoglobulini
Specifični humani imunoglobulini vsebujejo protitelesa proti določenim virusom in bakterijam. Zato jih lahko pripravimo iz mešanice omejenega števila enot.
Te zahteve izpolnjujejta naslednja specifična humana imunoglobulina:
humani imunoglobulin anti-tetanus,
humani imunoglobulin anti-vakcinija.
Mogoče je razviti tudi druge specifične imunoglobuline in ko bo zanje obstajal primeren mednarodni standard, jih bo treba določiti glede na ta standard in njihovo jakost izraziti v mednarodnih enotah.
Humani imunoglobulin anti-vakcinija vsebuje najmanj 500 IE na ml protiteles vakcinije, ki jih določimo z nevtralizacijskim testom na horioalantonskih membranah ali na tkivni kulturi. Humani imunoglobulin anti-tetanus vsebuje najmanj 50 IE na ml antitoksina tetanusa, ki ga določimo z nevtralizacijskim testom na živalih.
Specifični humani imunoglobulini morajo izpolnjevati tudi zahteve, opisane v razdelku 4, humani imunoglobulin.
Koncentracija imunoglobulina v končni raztopini je lahko med 100 in 170 g/l odvisno od vsebnosti protiteles.
Označevanje – Na etiketi vsebnika so navedeni vsi podatki, prikazani na vzorčni etiketi (Priloga 5). Na etiketi je navedena tudi jakost v mednarodnih enotah v skladu z ustreznim mednarodnim standardnim ali mednarodnim referenčnim pripravkom.
6. Dehidrirani humani fibrinogen
Dehidrirani humani fibrinogen je dehidrirani pripravek, ki vsebuje topno sestavino tekoče človeške plazme, ki po dodatku trombina preide v fibrin. Uporabljena metoda priprave je takšna, da dobimo snov, ki ustreza zahtevam, predpisanim v tem protokolu, in s katero je najmanjše tveganje prenosa serumskega hepatitisa. Mešanica plazme, ki jo uporabimo za pripravo fibrinogena, naj vsebuje čim manj enot.
Med pripravo ne dodamo nobene antiseptične ali bakteriostatične učinkovine. Končni pripravek liofiliziramo.
Topnost – Dodamo vodo do oznake; dehidrirani pripravek mora biti popolnoma topen. V 60 minutah po rekonstituciji se precipitacija ne sme pojaviti.
Identifikacija
i) Precipitacijski testi s specifičnimi antiserumi morajo pokazati le vsebnost proteinov človeške plazme.
ii) Sveže rekonstituirani pripravek ima po dodatku trombina lastnost koagulacije. Kadar trombin dodamo raztopini humanega fibrinogena enake koncentracije, kot je v sveži normalni plazmi, koagulacija nastopi v največ dvakratnem času, ki je potreben, da koagulacija po dodatku trombina nastopi v sveži normalni plazmi.
iii) Protein, ki koagulira. S trombinom koagulira najmanj 50 % vsega proteina.
Izguba mase pri dehidraciji – Pripravki ne smejo izgubiti več kot 0,5 % svoje mase po dodatnem 24-urnem sušenju ob prisotnosti fosforjevega pentoksida pri tlaku, ki ne presega 0,02 mm živega srebra.
Sterilnost – Po rekonstituciji je končni pripravek sterilen, ko ga preizkusimo z ustrezno bakteriološko metodo.
Shranjevanje -Humani fibrinogen hranimo v atmosferi dušika ali v vakuumu v sterilnem vsebniku, ki je zaprt tako, da se prepreči prodiranje mikroorganizmov, in kolikor je mogoče, vlage, zaščiten mora biti pred svetlobo in shranjen pri priporočeni temperaturi.
Označevanje – Na etiketi vsebnika so navedeni vsi podatki, prikazani na vzorčni etiketi (Priloga 6). Datum priprave je datum, ko ga damo v končno raztopino pred liofilizacijo.
7. Dehidrirani ali zamrznjeni humani koagulacijski faktor VIII
I. Zahteve, ki se nanašajo na krvodajalce
Krvodajalci morajo biti dobrega zdravja in zlasti ne smejo imeti nobene prenosljive bolezni v skladu z merili, sprejetimi za dehidrirano človeško plazmo.
II. Zahteve, ki se nanašajo na pripravke
Sterilnost in netoksičnost – Končni pripravek mora biti sterilen in apirogen. Kadar izvajamo krioprecipitacijo v plastičnih vrečkah, pripravek ne sme vsebovati organskega topila ali drugih tujih učinkovin, prisotnih v zamrzovalni tekočini. Prodiranje navedenih učinkovin skozi stene plastične vrečke lahko preprečimo, tako da pred zamrzovanjem damo vrečko v drugo neprepustno vrečko. Tveganje, da bi se plastična vrečka med hranjenjem v zamrznjenem stanju raztrgala, lahko zmanjšamo tako, da posamezno vrečko hranimo v zaščitni škatli.
Eritrociti, levkociti in trombociti – Pripravek centrifugiramo tako, da izločimo celične elemente čim bolj popolno in čim prej po odvzemu.
Topnost – Dodatek navedene količine ustreznega topila mora povzročiti, da se dehidrirani pripravek popolnoma raztopi v manj kot 30 minutah pri 37 C. Majhni in lahko ločljivi agregati fibrinogena so lahko še prisotni.
Stabilnost – Pripravek, ki smo ga raztopili in hranili tri ure pri 20 C, ne sme kazati nobenih znakov precipitacije.
Jakost – Rekonstituirani pripravek mora vsebovati navedeno najmanjšo količino faktorja VIII, ena enota ustreza jakosti 1 ml povprečne normalne sveže plazme; jakost določimo z metodo, ki jo odobri pristojni državni organ.
Odsotnost iregularnih protiteles – Če je pripravek namenjen bolnikom s katero koli krvno skupino AB0, titer protiteles anti-A in anti-B ne sme presegati 32.
Identifikacija – Precipitacijski testi s specifičnimi antiserumi morajo pokazati le vsebnost proteinov človeške plazme.
Izguba mase pri dehidraciji – Liofilizirani pripravki ne smejo izgubiti več kot 1,5 % svoje mase po dodatnem 24-urnem sušenju ob prisotnosti fosforjevega pentoksida pri tlaku, ki ne presega 0,02 mm živega srebra.
Shranjevanje – Humani faktor VIII hranimo v globoko zamrznjenem stanju pri temperaturi pod –30 C in v liofoliziranem stanju pri temperaturi pod 5 C, tako da je zaščiten pred svetlobo. Dehidrirani pripravek hranimo v atmosferi dušika ali v
vakuumu v sterilni viali, zaprti tako, da se prepreči prodiranje mikroorganizmov, in kolikor je mogoče, prodiranje vlage. Hranjenje v zamrznjenem stanju ne sme trajati več kot šest mesecev, v dehidriranem stanju eno leto, če pripravek ni bil ponovno testiran na najmanjšo zahtevano jakost.
III. Označevanje
Na etiketi pripravka so navedeni vsi podatki, ki so prikazani na vzorčni etiketi (Priloga 7).
8. Dehidrirani humani koagulacijski faktor IX
I. Zahteve, ki se nanašajo na krvodajalce
Krvodajalci morajo biti dobrega zdravja in zlasti ne smejo imeti nobene prenosljive bolezni v skladu z merili, sprejetimi za dehidrirano človeško plazmo.
II. Zahteve, ki se nanašajo na koncentrat
Sterilnost in netoksičnost – Končni pripravek, ki je bil testiran z ustreznimi metodami, mora biti sterilen, apirogen in ne sme imeti neželenih vazodepresornih ali respiratornih učinkov. Test na odsotnost vazodepresornih učinkov je treba izvesti na psu ali mački.
Topnost – Dodatek navedene količine topila mora povzročiti, da se dehidrirani pripravek pri 37 C v 10 minutah popolnoma raztopi.
Aktivnost tromboplastina in odsotnost prostega trombina – Rekalcifikacijski čas normalne plazme, merjen pri 37 C v prisotnosti enakega volumna različnih razredčenih raztopin rekonstituiranega pripravka, ne sme biti krajši od 40 sekund. Rekonstituirani pripravek, ki smo mu dodali enak volumen fibrinogena (3 g/l), pri temperaturi 37 C ne sme koagulirati v 6 urah.
Jakost – Rekonstituirani pripravek mora vsebovati navedeno najmanjšo količino faktorja XI, ena enota ustreza jakosti 1 ml povprečne normalne sveže plazme; jakost določimo z metodo, ki jo odobri pristojni državni organ.
Vsebnost in stabilnost in vivo – Metoda za pripravo pripravka mora biti izbrana tako, da hitra intravenozna aplikacija 50 enot na kg telesne teže s pomočjo različnih serij snovi, ki jih damo več bolnikom, v odsotnosti specifičnega inhibitorja in v bazalnih pogojih v 15 minutah povzroči povprečen porast najmanj 300 enot na liter plazme in po trajanju učinka po 24 urah povprečen porast najmanj 60 enot na liter plazme.
Identifikacija – Precipitacijski testi s specifičnimi antiserumi morajo pokazati izključno vsebnost proteinov človeške plazme.
Izguba mase pri dehidraciji – Pripravek ne sme izgubiti več kot 1,5 odstotka svoje mase po dodatnem 24-urnim sušenju ob prisotnosti fosforjevega pentoksida pri tlaku, ki ne presega 0,02 mm živega srebra.
Shranjevanje – Dehidrirane pripravke moramo hraniti pri temperaturi pod 5 C. Pripravka ne smemo hraniti več kot dve leti, če njegova jakost ni bila ponovno testirana.
III. Označevanje
Na etiketi pripravka so navedeni vsi podatki, ki so prikazani na vzorčni etiketi (Priloga 8).
PRILOGA 1 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
Certifikat
(4. člen)
NE SME SE LOČITI OD POŠILJKE
.............................................19..........
(kraj) (datum)
Število paketov Podpisani potrjuje, da je pošiljka, opisana ob robu,
......................... ..............................................................................................
za katere pripravo je odgovoren.......................................
Oznake ..............................................................................................
......................... ..............................................................................................
......................... eden od organov, navedenih v 6. členu sporazuma;
Številka serije skladna s specifikacijami protokola k sporazumu,
in se lahko takoj dostavi prejemniku (ime in kraj)
.......................... ...............................................................................................
(žig) (podpis) (naziv)
PRILOGA 2 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Polna človeška kri
3. Referenčna številka:
4. Krvna skupina:
5. Krvna skupina Rh:
6. ... ml antikoagulantne raztopine
... g/l glukoze
... M dinatrijevega citrata
... ml krvi
7. Titer izohemolizinov določen
ni določen
8. Datum odvzema:
Rok uporabnosti:
9. Hraniti pri 4 C do 6 C.
10. Ne sme se uporabiti, če so vidni znaki sprememb pripravka.
PRILOGA 2 bis K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Koncentrirani človeški eritrociti:
3. Referenčna številka:
4. Krvna skupina:
5. Krvna skupina Rh:
6. ... ml pripravljeno iz ... ml krvi
7. Volumen in sestava uporabljenega antikoagulanta:
8. Datum odvzema:
Datum priprave:
Rok uporabnosti:
9. Hraniti pri 2 C do 6 C.
10. Dodana ohranitvena raztopina: volumen
sestava
PRILOGA 3 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Dehidrirana človeška plazma:
3. Referenčna številka:
4. Rekonstituirati z ... ml sterilne, apirogene destilirane vode.
5. Rekonstituirana plazma vsebuje:
... g/l glukoze
... M dinatrijevega citrata
... g/l koncentracije proteinov (najmanj)
6. Število posameznih odvzemov v mešanici:
7. Datum priprave:
Rok uporabnosti:
8. Hraniti zaščiteno pred svetlobo pod 20 C.
9. Uporabiti takoj po rekonstituciji.
PRILOGA 4 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Dehidrirani humani albumin:
3. Številka serije:
4. Albumin ... g
Stabilizator, vrsta .., ... g/l v rekonstituirani raztopini
Natrij ... mM/g albumina
5. Datum priprave:
Rok uporabnosti:
6. Rekonstituirati z ... ml sterilne, apirogene destilirane vode.
7. Hraniti zaščiteno pred svetlobo pod 20 C.
8. Uporabiti takoj po rekonstituciji.
PRILOGA 4 (1. nadaljevanje)
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Raztopina humanega albumina
3. Številka serije:
4. Albumin ... g/l
5. Stabilizator, vrsta ... , ... g/l
6. Natrij: ... mM/g albumina8. Datum priprave:
Rok uporabnosti:
9. Hraniti zaščiteno pred svetlobo pri 4 C do 6 C.
10. Sme se uporabiti le, če je raztopina bistra in brez usedlin.
PRILOGA 4 (2. nadaljevanje)
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Proteinska frakcija človeške plazme ... ml:
3. Številka serije:
4. Albumin ... g/l
Stabilizator, vrsta ..., ... g/l
Natrij: ... mM/g5. Datum priprave:
Rok uporabnosti:
6. Hraniti zaščiteno pred svetlobo pri 4 C do 6 C.
7. Sme se uporabiti le, če je raztopina bistra in brez usedlin.
PRILOGA 5 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Humani imunoglobulin:
3. Številka serije:
4. Skupaj proteinov ... g/l
Druge dodane snovi, vrsta ..., ... g/l
Skupni volumen ... ml
5. Datum priprave:
Rok uporabnosti:6. Hraniti zaščiteno pred svetlobo pri 4 C do 6 C.
7. Ni za intravenozno uporabo.
PRILOGA 6 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Dehidrirani humani fibrinogen:
3. Številka serije:
4. Protein, ki koagulira ... g
Druge dodane snovi, vrsta ..., ... g/l rekonstituirane raztopine
5. Datum priprave:
Rok uporabnosti:
6. Rekonstituirati z ... ml sterilne, apirogene destilirane vode.
7. Število posameznih enot v mešanici: ...
8. Hraniti zaščiteno pred svetlobo pod 20 C.
9. Uporabiti takoj po rekonstituciji.
PRILOGA 7 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Zamrznjeni humani koagulacijski faktor VIII ali
dehidrirani humani koagulacijski faktor VIII:
Metoda priprave:
3. Številka serije:
4. Najmanjša količina faktorja VIII, količina vseh proteinov, vrsta in količina
dodane učinkovine:
5. Vrsta in volumen topila:
6. Število krvodajalcev na serijo:
7. Titer hemaglutininov ni večji od 1 : 32 ali
krvna skupina AB0
8. Datum priprave:
9. Rok uporabnosti:
10. Hraniti zaščiteno pred svetlobo in zamrznjeno pod –30 C
ali v dehidriranem stanju pri temperaturi pod 5 C.
11. Po rekonstituciji uporabiti intravenozno takoj ali najpozneje po 3 urah
hranjenja pri 20 C.
PRILOGA 8 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Dehidrirani humani koagulacijski faktor IX:
Drugi prisotni koagulacijski faktorji:
Metoda priprave:
3. Številka serije:
4. Najmanjša količina faktorja IX, količina vseh proteinov, vrsta in količina
dodane učinkovine:
5. Vrsta in volumen topila:
6. Število krvodajalcev na serijo:
7. Datum priprave:
8. Rok uporabnosti:
9. Hraniti zaščiteno pred svetlobo pod 5 C.
10. Po rekonstituciji pripravka takoj uporabiti intravenozno.
PRILOGA 9 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Sterilna, apirogena destilirana voda
Za rekonstitucijo dehidrirane človeške plazme
dehidriranega humanega albumina
dehidriranega humanega fibrinogena
ali dehidriranih humanih koagulacijskih faktorjev VIII in IX
3. Količina ... ml
PRILOGA 10 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
1. Ime in naslov proizvajalca:
2. Set za dajanje
Set za dajanje polne človeške krvi, rekonstituirane dehidrirane človeške plazme, humanega albumina, proteinske frakcije človeške plazme, humanega fibrinogena in dehidriranega ali zamrznjenega koagulacijskega faktorja VIII ali dehidriranega humanega koagulacijskega faktorja IX.
PRILOGA 11 K PROTOKOLU
SVET EVROPE
Evropski sporazum o izmenjavi
zdravilnih učinkovin človeškega izvora
NETOKSIČNOST PLASTIČNE OPREME
ZA TRANSFUZIJO KRVI
I. Kemični testi
Testi so namenjeni za preizkušanje plastične opreme za transfuzijo krvi. To opremo
sestavljajo:
1. plastični vsebniki za zbiranje, ločevanje in hranjenje krvi in krvnih pripravkov,
2. plastični seti za odvzem in dajanje krvi.
Material preizkusimo, ko je že bil steriliziran z metodo končne sterilizacije. Preizkušamo:
1. plastiko, ki se uporablja za izdelavo vsebnikov,
2. cevke na vsebnikih,
3. set za odvzem in dajanje krvi.
Vsebnike preizkusimo, preden jih napolnimo z antikoagulantno raztopino. Če vsebnike preizkusimo, potem ko so že napolnjeni z antikoagulantno raztopino, pri vrednotenju rezultatov testov, izvedenih na vsebnikih, upoštevamo v razdelku III navedene teste mejnih vrednosti, ki so bili izvedeni na antikoagulantni raztopini.
Pristojni zdravstveni organ zahteva, da mu izdelovalec opreme za transfuzijo razkrije podrobno sestavo plastičnega materiala ali materialov in drugih snovi, ki jih uporablja pri izdelavi opreme, izvor sestavin materiala ali materialov in njihov način izdelave (oziroma referenčne številke sestavin), podrobne podatke o izdelavi opreme, vrsti dodatkov in veziv, če jih dodaja med predelavo, in metodo sterilizacije. Izdelovalec pri vsem omenjenem ne sme ničesar spremeniti brez predhodne predložitve spremembe ustreznemu zdravstvenemu organu in njegove odobritve.
Vsaka serija surovine, ki se uporablja za izdelavo opreme, mora biti opredeljena s serijsko številko, ki jo izdelovalec opreme zapiše skupaj z identifikacijskimi številkami vseh serij opreme za transfuzijo, izdelanih iz teh surovin, in z rezultati vseh testov, ki se nanašajo na te serije.
Izvedeni morajo biti vsi možni previdnostni ukrepi, da se zmanjša tveganje naključne kontaminacije med posameznimi fazami proizvodnega procesa.
A. Priprava ekstrakta in slepega vzorca
a) Za spodaj opisani test potrebujemo 1250 cm2 plastike (celotna površina, obe strani, vzorca plastike v obliki folije s površino 625 cm2). Vzorec – brez vsakega tiska ali etiket – razrežemo na kose s površino največ 10 cm2.
Dolžino (L) cevk v cm izračunamo takole:
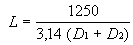
D1 = notranji premer v cm
D2 = zunanji premer v cm
Cevke režemo vzdolžno v približno 10 cm dolge delce. Za ekstrakcijo uporabimo
10 ml vode na površini 50 cm2.
b) Delce plastične folije ali cevi damo v vsebnik iz borosilikatnega stekla z 250 ml apirogene destilirane vode, pridobljene iz učinkovitega destilatorja s steklenimi kondenzacijskimi površinami in zbiralnimi cevkami1. Odprtino vsebnika pokrijemo z obrnjeno čašo, vsebnik pa nato 30 minut segrevamo v nasičeni pari pri 110 C (avtoklaviranje), nato pa ga hitro ohladimo na sobno temperaturo in z apirogeno destilirano vodo volumen dopolnimo do 250 ml. Pri tem ni pomembno, če se plastični vzroci rahlo sprijemajo.
Plastične materiale, ki so občutljivi za vročino, lahko namesto segrevanja v avtoklavu segrevamo pri 70 C 72 ur.
Slepi vzorec pripravimo na podoben način brez plastike.
_________________________________
1 Če je bila plastika v stiku z antikoagulantno raztopino, kose najprej damo v podoben vsebnik z mrzlo destilirano vodo (100 ml) in jih večkrat pretresemo. To enkrat ponovimo.
B. Testiranje ekstrakta
1. Oksidirajoča snov
V Erlenmeyerjevi bučki iz borosilikatnega stekla k 20 ml ekstrakta dodamo 20 ml
2 mM kalijevega hipermanganata in 0,1 ml 1M žveplene kisline; zmes naj vre 3 minute. Raztopino hitro ohladimo in dodamo 0,1 g kalijevega jodida in 5 kapljic škrobove raztopine. Titriramo z raztopino, ki vsebuje 10 mM natrijevega tiosulfata. Hkrati izvedemo tudi titracijo slepega vzorca. Razlika v volumnu tiosulfata, ki smo ga uporabili za obe titraciji, naj ne presega 2,00 ml raztopine 10 mM natrijevega tiosulfata.
2. Klorid
Ekstrakt ustreza testu mejne vrednosti za klorid, kadar je količina klorida največ
1,12 M klorida.
3. Amoniak
Ekstrakt ustreza testu mejne vrednosti za amoniak, kadar je količina NH3 največ
120 M NH3.
4. Fosforna kislina – fosfat
Ekstrakt ustreza testu mejne vrednosti za fosfat.
Mejni test na fosfate
25 ml ekstrakta v Kjeldahlovi bučki skoraj do suhega izparevamo, ohladimo ostanek, dodamo 2 kapljici žveplene kisline in 1 ml dušikove kisline, zmes segrevamo, dokler se ne pojavijo beli hlapi, nato jo ohladimo. Dodamo 1 kapljico perklorove kisline in pol ure zmerno segrevamo. Ohladimo ostanek in dodamo vodo do oznake 25 ml.
10 ml raztopine prenesemo v 25-mililitrsko titrirno bučko, dodamo 8 ml raztopine amoniakalnega molibdata žveplene kisline in 2 ml sveže pripravljene raztopine askorbinske kisline s koncentracijo 100 g/l. 30 minut segrevamo v vodni kopeli pri
50 C, ohladimo in razredčimo zmes do oznake 25 ml. Zelena ali modra barva raztopine ni intenzivnejša od barve, ki smo jo dobili z enako obdelavo 25 ml slepega vzorca.
5. Kislost ali bazičnost
10 ml ekstrakta, ki mu dodamo 2 kapljici raztopine fenolftaleina, se ne sme obarvati rdeče; dodati smemo največ 0,4 ml raztopine, ki vsebuje 10 mM natrijevega hidroksida, da dobimo rdečo barvo. Ko barvo odstranimo z dodatkom 0,08 ml
10 mM solne kisline z dodatkom 5 kapljic raztopine metil rdečega, dobimo rdečo ali oranžnordečo barvo.
6. Ostanek po izparevanju
100 ml ekstrakta izparevamo v vodni kopeli do suhega in osušimo pri 105 C do stalne mase. Ostanek tehta največ 5,0 mg.
7. Bistrost in barva
Ko ga primerjamo s slepim vzorcem, je ekstrakt debeline 5 cm bister in brezbarven.
8. Okus in vonj
V primerjavi s slepim vzorcem je ekstrakt brez vonja in okusa.
9. Posebni elementi
Ekstrakt ustreza mejnim testom na
i) kateri koli naslednji element: arzen, krom, baker, svinec, silicij, srebro in
kositer, kadar je njihova količina enaka 1g/ g,
ii) kadmij, če je njegova količina enaka 0,1 g/g.
10. Ostanek po žarjenju
Ko 1,0 g plastičnega materiala žarimo do stalne mase, ostane največ 1 mg ostanka.
11. Težke kovine
Ostanek po žarjenju raztopimo v najmanjši količini raztopine 2 M solne kisline in segrejemo, če je potrebno. Izvedemo ustrezni mejni test na težke kovine. Plastični material ustreza, če ne presegamo mejne vrednosti 5 g/g, preračunano na svinec.
II. Biološki testi
1. Test na netoksičnost izvedemo pri začetnem vrednotenju sestave plastike, ki je namenjena izdelavi vsebnikov ter setov za odvzem in dajanje s pomočjo ekstrakta A, in po vsaki novi seriji materialov z odobreno sestavo s pomočjo ekstrakta B po postopku, natančno navedenem v državni farmakopeji, ali po kaki drugi metodi, ki jo odobri državni kontrolni organ. (Ekstrakta A in B sta opredeljena v opombi spodaj.)
2. Test na apirogenost izvedemo pri začetnem vrednotenju sestave plastike, ki je namenjena izdelavi vsebnikov ter setov za odvzem in dajanje s pomočjo ekstrakta A, in po vsaki novi seriji materialov z odobreno sestavo s pomočjo ekstrakta C in pri običajni kontroli vsebnikov in setov s pomočjo ekstrakta C po postopku, natančno navedenem v državni farmakopeji, ali po kaki drugi metodi, ki jo odobri državni kontrolni organ.
Pogostnost testiranja pirogenov s pomočjo ekstrakta C določi državni kontrolni organ.
(Ekstrakta A in C sta opredeljena v opombi spodaj.)
3. Test na hemolitične učinke v puferskih sistemih izvedemo pri začetnem vrednotenju sestave plastike, ki
je namenjena izdelavi vsebnikov ter setov za odvzem in dajanje, in po vsaki novi seriji materialov z odobreno sestavo s pomočjo ekstrakta, opisanega v razdelku I A zgoraj. (Glede metode in sprejemljive mejne vrednosti glej dodatek k tej prilogi.)
4. Test na preživetje eritrocitov in vivo izvedemo pri začetnem vrednotenju sestave plastike, ki je namenjena izdelavi vsebnikov za kri. Če je v dogovorjeni sestavi kakšna sprememba, test ponovimo. (Glede predlaganih metod in sprejemljivih mejnih vrednosti glej dodatek k tej prilogi.)
Opomba
Ekstrakt A pripravimo tako, da ekstraktu, opisanem pod I A zgoraj, dodamo apirogeni natrijev klorid do končne koncentracije 9 g/l.
Ekstrakt B:
Set za transfuzijo. Set za transfuzijo v celoti napolnimo s sterilno apirogeno raztopino, ki vsebuje 9 g/l natrijevega klorida, s stiščkoma dobro stisnemo konca in napolnjeni set za eno uro potopimo v vodo s stalno temperaturo 85 C. Zberemo vsebino iz seta.
Plastični vsebnik. Če je vsebnik napolnjen z antikoagulantno raztopino, ga izpraznimo in pri temperaturi 20 C dvakrat izperemo s po 250 ml sterilne apirogene destilirane vode. Vsebnik napolnimo s 100 ml sterilne apirogene raztopine, ki vsebuje
9 g/lnatrijevega klorida, ga dobro zapremo in v vodoravnem položaju za eno uro potopimo v vodo s stalno temperaturo 85 C. Zberemo vsebino iz vsebnika.
Ekstrakt C:
Set za transfuzijo. Pri sobni temperaturi spustimo s hitrostjo pretoka približno 10 ml na minuto skozi najmanj deset setov po 40 ml sterilne apirogene raztopine natrijevega klorida s koncentracijo 9 g/l. Zbrano raztopino testiramo.
Plastični vsebnik. Prazen. Pri sobni temperaturi spustimo skozi zbiralne cevke najmanj štirih plastičnih vsebnikov po 100 ml sterilne apirogene raztopine, ki vsebuje 9 g/l natrijevega klorida. Raztopina naj ostane v vsebniku deset minut; zbiramo tekočine, ki pri izpraznjenju odtečejo skozi iztočne cevke. Zbrano raztopino testiramo.
Plastični vsebnik z antikoagulantom (glej razdelek III).
DODATEK
BIOLOŠKI TEST – MEJNE VREDNOSTI IN METODE
A. Test na netoksičnost
(Glej postavko II točko 1) priloge zgoraj): mejna vrednost, kot je določena v državni farmakopeji.
B. Test na apirogenost
(Glej postavko II točko 2) priloge zgoraj): mejna vrednost, kot je določena v državni farmakopeji.
C.Test na hemolitične učinke v puferskih sistemih
(Glej postavko II točko 3) priloge zgoraj):
a) Mejna vrednost:
Kar zadeva elektrolitsko ozmotsko delovanje, slana raztopina, ki vsebuje 5,0 g/l NaCl, ne sme povzročiti hemolize, katere vrednost bi bila večja od 10 %, in hemolizna vrednost slane raztopine s koncentracijo 4,0 g/l se ne sme razlikovati za več kot 10 % od vrednosti, ki jo povzroči ustrezna kontrolna raztopina.
b) Metoda:
Iz primarne puferske osnovne raztopine za hemolizo pripravimo tri raztopine: 30 ml puferske osnovne raztopine in 10 ml vode (raztopina a0), 30 ml puferske osnovne raztopine in 20 ml vode (raztopina b0) in 15 ml puferske osnovne raztopine in 85 ml vode (raztopina c0).
V vsako od treh epruvet za centrifugiranje (1, 2 in 3) dodamo 1,40 ml ekstrakta. V epruveto 1 dodamo 0,10 ml a0, v epruveto 2 0,10 ml b0 in v epruveto 3 0,10 ml c0, da dobimo slane raztopine, enakovredne raztopinam, ki vsebujejo 5,0 (epruveta 1), 4,0 (epruveta 2) in 1,0 g/l NaCl (epruveta 3), kar zadeva elektrolitsko ozmotsko delovanje. V vsako epruveto dodamo 20 l sveže, dobro premešane, heparinizirane človeške krvi. Epruvete za 40 minut postavimo v vodno kopel s temperaturo 30 C
( 1 C). Tri raztopine, ki vsebujejo 3,0 ml a0 in 12,0 ml vode (raztopina a1), 4,0 ml b0 in 11,0 ml vode (raztopina b1) in 4,75 ml b0 in 10,25 ml vode (raztopina c1), so tako pripravljene.
V prvo epruveto dodamo 1,50 ml a1, v drugo 1,50 ml b1 in v tretjo 1, 50 ml c1, centrifugiramo 5 minut pri 2.000 do 2.500 obratih na minuto v centrifugi s pomičnimi kivetami.
Hkrati za vsako od koncentracij pripravimo kontrolne raztopine, v katerih ekstrakt nadomestimo z vodo.
Pri 540 nm izmerimo ekstinkcijo tekoče plasti. Kot pripravek za slepi preizkus uporabimo pufersko osnovno raztopino za hemolizo. Vrednost hemolize v odstotku izračunamo po naslednji formuli:
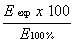
kjer je E100 % = ekstinkcija raztopine, ki vsebuje ekvivalent 1,0 g/l soli,
in Eexp = ekstinkcija raztopin, ki vsebujejo ekvivalent 4,0 oz. 5,0 g /l soli.
Puferska osnovna raztopina za hemolizo
V destilirani vodi raztopimo 90,0 g natrijevega klorida, 13,7 g anhidričnega dinatrijevega fosfata in 1,90 g anhidričnega mononatrijevega fosfata ter dodamo destilirano vodo do oznake 1000,0 ml.
D. Test na preživetje eritrocitov in vivo
(Glej postavko II točko 4) priloge zgoraj)
a) Mejna vrednost:
Od eritrocitov v polni človeški krvi z antikoagulantom ACD, ki je bila shranjena
21 dni pri 4 C do 6 C, jih mora najmanj 70 % preživeti 24 ur po transfuziji. To lahko določimo po eni izmed metod, navedenih pod b) spodaj.
b) Predlagane metode:
1. Metoda ISO /TC/ 76 /WGD / 3, Method of ISO/TC/76/WGD/3, App. E.
2. Ashbyjeva metoda, Ashby Technique - Ashby, W. The determination of the length of life of transfused blood corpuscles in man.
J. Exp. Med. 29: 267 - 82, 1919.
Young L. E. Platzer, R. F. in Rafferty, J. A. Differential agglutination of human
erythrocytes.
J. Lab. Clin. Med. 32: 489 - 501, 1947.
3. Gibson-Scheitlinova metoda, The Gibson-Scheitlin method - Gibson, J. G. in Scheitlin, W. A. A method employing radio-active chromium for assaying the viability of human erythrocytes returned to the circulation after refrigerated storage.
J. Lab. Clin. Med. 46: 679 - 88, 1955.
4. Strumijeva metoda, The Strumia method - Strumia, M. M., Taylor, L., Sample
A. B., Colwell, L. S. and Dugan, A. Uses and limitations of survival studies of erythrocytes tagged with Cr 51.
Blood 10 : 429 - 40, 1955.
5. Metoda Cr51–J125, Cr51–J125 technique - Button, L. N., Gibson. J. G. in Walter,
C. W. Simultaneous determination of the volume of red cells and plasma for survival studies of stored blood.
Transfusion 5: 143 - 148, 1965.
6. Priporočena metoda za radioizotopsko določanje preživetja eritrocitov, Recommended Method for Radioisotope Red Cell Survival studies, Brit. J. Haemat. 21 : 242, 1971.
III. Zahteve za antikoagulantno raztopino
v plastičnih vsebnikih
Vsak vsebnik vsebuje količino in sestavo antikoagulantne raztopine, navedene na etiketi, za volumen krvi, ki bo odvzeta.
Antikoagulantna raztopina in/ali sestavine, uporabljene pri njeni pripravi,
izpolnjuje zahteve državne farmakopeje posamezne države.
Antikoagulantna raztopina izpolnjuje zahteve državne farmakopeje posamezne države glede mejnih vrednosti za težke kovine, odsotnosti delcev snovi, netoksičnosti in apirogenosti.
Sklenjeno v Strasbourgu 19. aprila 1982.
Dodatni protokol k
Evropskemu sporazumu
o izmenjavi
zdravilnih učinkovin
človeškega izvora
Države članice Sveta Evrope, pogodbenice Evropskega sporazuma o izmenjavi zdravilnih učinkovin človeškega izvora z dne 15. decembra 1958 (v nadaljevanju "sporazum"), so se
ob upoštevanju določb prvega odstavka 5. člena sporazuma, po katerem "pogodbenice ukrenejo vse potrebno, da so zdravilne učinkovine človeškega izvora, ki jim jih dajo na voljo druge pogodbenice, oproščene vseh uvoznih dajatev",
glede na to, da je za odobritev te oprostitve državam članicam Evropske gospodarske skupnosti pristojna Skupnost, ki je za to pooblaščena na podlagi pogodbe, s katero je bila ustanovljena,
glede na to, da je za izvajanje prvega odstavka 5. člena sporazuma potrebno, da Evropska gospodarska skupnost lahko postane pogodbenica sporazuma,
sporazumele o naslednjem:
1. člen
Evropska gospodarska skupnost lahko postane pogodbenica sporazuma, tako da ga podpiše. Za Skupnost začne sporazum veljati prvi dan v mesecu, ki sledi podpisu.
2. člen
1. Ta dodatni protokol je na voljo za sprejetje pogodbenicam sporazuma. Veljati začne prvi dan v mesecu, ki sledi dnevu, ko zadnja pogodbenica deponira svojo listino o sprejetju pri generalnem sekretarju Sveta Evrope.
2. Ne glede na to začne ta dodatni protokol veljati po izteku dveh let od dneva, ko je dan na voljo za sprejetje, razen če ena od pogodbenic uradno izjavi, da nasprotuje začetku veljavnosti. Če je bilo uradno obveščeno o takem nasprotovanju, se uporabi prvi odstavek tega člena.
3. člen
Od dneva začetka veljavnosti je ta dodatni protokol sestavni del sporazuma. Po tem dnevu ne more nobena država postati pogodbenica sporazuma, ne da bi hkrati postala pogodbenica dodatnega protokola.
4. člen
Generalni sekretar Sveta Evrope uradno obvesti države članice Sveta Evrope, vsako državo, ki je pristopila k sporazumu, in Evropsko gospodarsko skupnost o vsakem sprejetju ali nasprotovanju po 2. členu in o datumu začetka veljavnosti tega dodatnega protokola v skladu z 2. členom.
Generalni sekretar tudi Evropsko gospodarsko skupnost uradno obvesti o vsakem aktu, uradnem obvestilu ali sporočilu, ki se nanaša na sporazum.
Sestavljeno v Strasbourgu 29. septembra 1982 v angleškem in francoskem jeziku in dano na voljo za sprejetje 1. januarja 1983. Besedili sta enako verodostojni in se v enem izvodu hranita v arhivu Sveta Evrope. Generalni sekretar Sveta Evrope pošlje overjene kopije vsaki državi članici Sveta Evrope, vsaki državi, ki je povabljena, da pristopi k sporazumu, in Evropski gospodarski skupnosti.
3. člen
Za izvajanje sporazuma, protokola in dodatnega protokola skrbita Ministrstvo za finance in Ministrstvo za zdravstvo.
4. člen
Ta zakon začne veljati naslednji dan po objavi v Uradnem listu Republike Slovenije - Mednarodne pogodbe.
Številka: 500-02/00-3/1
Ljubljana, dne 19. julija 2000
Predsednik
Državnega zbora
Republike Slovenije
Janez Podobnik, dr. med.
| Zadnja sprememba: 08/09/2007 | Zbirke Državnega zbora RS - sprejeti zakoni |